
ABSTRACT
Wide-ranging activation of the innate immune system causing chronic low-grade inflammation is closely involved not only in the pathogenesis of type 2 diabetes mellitus and its complications, through an ongoing cytokine-induced acute-phase response, but also in the pathogenesis of periodontal diseases, whereby cytokines play a central role in the host’s response to the periodontal biofilm. Although there is extensive knowledge about the pathways through which diabetes affects periodontal status, less is known about the impact of periodontal diseases on the diabetes-related inflammatory state. This review attempts to explain the immunobiological connection between periodontal diseases and type 2 diabetes mellitus, exploring the mechanisms through which periodontal infection can contribute to the low-grade general inflammation associated with diabetes (thus aggravating insulin resistance) and discussing the impact of periodontal treatment on glycemic control in people living with both diabetes and periodontal disease.
Introduction
Diabetes mellitus is a clinically and genetically heterogeneous group of disorders affecting the metabolism of carbohydrates, lipids and proteins, in which hyperglycemia is a main feature. These disorders are due to a deficiency in insulin secretion caused by pancreatic β-cell dysfunction and/or insulin resistance in liver and muscle.1 Diabetes affects about 21 million people in the United States, or more than 9% of the adult population, and has a dramatic impact on the health care system through high morbidity and mortality among affected individuals.1 In Ontario, population-based data have revealed that the prevalence of diabetes increased by 69% over a recent 10-year period (from 5.2% in 1995 to 8.8% in 2005), which exceeded the global rate of increase of 39% that was predicted for the period 2000 to 2030.2 Furthermore, the rates of increase rose to a greater extent in the younger population. This increase was attributable to both a rise in incidence and a decline in mortality.2 Similarly, in the First Nations community of Kahnawake, Quebec, the prevalence rates of type 2 diabetes increased over the period 1986 to 2003, from 6.0% to 8.4% among males and from 6.4% to 7.1% among females.3
Type 1 diabetes results from cellular-mediated autoimmune destruction of pancreatic β-cells, which usually leads to total loss of insulin secretion; in contrast, type 2 diabetes is caused by resistance to insulin combined with a failure to produce enough additional insulin to compensate for the resistance.1 Type 2 diabetes is commonly linked to obesity, which contributes to insulin resistance through elevation of circulating levels of free fatty acids derived from the adipocytes; these free fatty acids inhibit glucose uptake, glycogen synthesis and glycolysis. In many obese individuals, insulin resistance is compensated by increased insulin production. However, in one-third of obese individuals, β-cell mass is reduced by a marked increase in β-cell apoptosis, which results in inadequate production of insulin.1
It seems that metabolic control is important not only in the pathogenesis and progression of the microvascular and macrovascular complications of diabetes mellitus,4 but also in the high susceptibility of these patients to infectious diseases, as evidenced by a 2- to 5-fold higher risk for periodontitis. Conversely, the risk of periodontitis is reduced by effective control of hyperglycemia.5
Several biologically plausible mechanisms have been proposed to explain the interactions between diabetes and periodontal diseases. These potential mechanisms are strikingly similar to those associated with the established complications of chronic diabetes, which suggests that periodontitis should be considered the sixth “classic” complication of diabetes.6 Less clear is the impact of periodontal diseases on glycemic control in diabetes and the mechanisms through which this effect might occur. It has been proposed that type 2 diabetes is a manifestation of the host’s inflammatory response, because an ongoing cytokine-induced acute-phase response is closely involved in the pathogenesis of this disease and its associated complications, such as dyslipidemia and atherosclerosis7 (Fig. 1). This cytokine-induced response is a low-grade inflammation that occurs through activation of the innate immune system. Increased serum concentrations of acute-phase response markers and cytokines have been observed in patients with type 2 diabetes, which indicates that circulating inflammatory cytokines modify the risk for type 2 diabetes.8 Likewise, the mechanisms of the host-mediated response in periodontal disease involve activation of the broad axis of innate immunity, specifically by upregulation of proinflammatory cytokines from monocytes and polymorphonuclear leukocytes, in the presence of gram-negative subgingival bacterial biofilm.9 Thus, chronic gram-negative periodontal infections may induce or perpetuate an elevated chronic systemic inflammatory state, contributing to increased insulin resistance and poor glycemic control1,10 (Fig. 2).
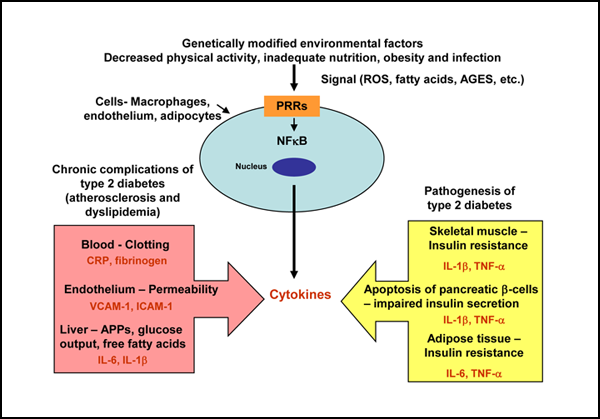
Figure 1: Innate immunity and type 2 diabetes mellitus. Cell components of the innate immune system, such as macrophages, endothelial cells and adipocytes, detect, through pattern-recognition receptors (PRRs), potential environmental threats to the host, which are represented by signals such as reactive oxygen species (ROS), fatty acids and advanced glycation end products (AGES). This process activates nuclear transcription factors, such as nuclear factor-kappa B (NF-κB), which induce immune inflammatory genes, which in turn cause the release of cytokines. These cytokines act in many cells in the body to produce the clinical and biochemical features of type 2 diabetes and its chronic complications. APPs = acute-phase proteins, CRP = C-reactive protein, IL = interleukin, TNF-α = tissue necrosis factor alpha, VCAM-1 = vascular cell adhesion molecule 1, ICAM-1 = vascular endothelial growth factor expression of intercellular adhesion molecule 1.

Figure 2: Innate immunity, periodontitis and type 2 diabetes mellitus. Periodontal diseases also involve activation of the broad axis of innate immunity through upregulation of proinflammatory cytokines from monocytes and polymorphonuclear leucocytes, including interleukin (IL)-1β, IL-6, IL-8, tumour necrosis factor alpha (TNF-α) and prostaglandin E2. Inappropriate secretion of these cytokines, in terms of either type or quantity, characterizes a dysregulated immune response that leads to destruction of periodontal tissues in the presence of gram-negative bacterial biofilm. These locally produced cytokines move into the systemic circulation, where they may perpetuate an elevated inflammatory state, worsening the patient's diabetes through increasing insulin resistance and glucose levels. AGES = advanced glycation end products, LPS = lipopolysaccharide.
This review discusses the relationship between periodontitis and type 2 diabetes mellitus, focusing on the mechanisms through which periodontal infections contribute to the diabetes-related inflammatory state, the influence of periodontal infections on insulin resistance and the ways in which treatment of these infections can influence glycemic control.
Mechanisms by Which Periodontitis May Influence Diabetes-Related Inflammatory State and Insulin Resistance
Evidence has consistently indicated that diabetes is a risk factor for increased severity of gingivitis and periodontitis.1,11 Conversely, periodontitis may be a risk factor for worsening glycemic control among patients with diabetes and may increase the risk of diabetic complications. Periodontitis may initiate or propagate insulin resistance in a manner similar to that of obesity, by enhancing activation of the overall systemic immune response initiated by cytokines (Fig. 3).11,12

Figure 3: Proposed mechanism by which periodontal inflammatory mediators may contribute to the development of insulin resistance in individuals with both type 2 diabetes and periodontitis. The inflammatory mediators originating from periodontal sources can interact systemically with lipids, free fatty acids and advanced glycation end products (AGES), all of which are characteristic of diabetes. This interaction induces or perpetuates activation of the intracellular pathways, such as the I-kappa-B (IκB), I-kappa-B kinase-β (IKKβ), nuclear factor-kappa B (NF-kβ) and the protein c-Jun N-terminal kinase (JNK) axes, all of which are associated with insulin resistance. The activation of these inflammatory pathways in immune cells (monocytes or macrophages), endothelium cells, adipocytes, hepatocytes and muscle cells promotes and contributes to an increase in the overall insulin resistance, which makes it difficult to achieve metabolic control in patients with both type 2 diabetes and periodontitis. IL = interleukin, IRS-1 = insulin receptor substrate-1, LPS = lipopolysaccharide, PGE2 = prostaglandin E2, PKCs = protein kinases C, PRRs = pattern-recognition receptors, pS302 (serine-302) and pS307 (serine-307) = examples of serine sites, ROS = reactive oxygen species, TNF-α = tumour necrosis factor alpha.
Findings from the Third National Health and Nutrition Examination Survey (NHANES III) in the United States13 showed that the prevalence of diabetes among people with periodontal disease (n = 1293) was 12.5%, whereas only 6.3% of periodontally healthy participants (n = 12 178) reported that they had diabetes, a 2-fold difference. Other studies have shown an association between the severity of periodontitis and glucose intolerance, signs of metabolic syndrome and additional diabetes-related complications, such as cardiovascular problems.14-16
There is limited knowledge about the mechanisms through which periodontal diseases may influence the diabetic state. In untreated severe periodontal disease, the cumulative surface area of ulcerated pocket epithelium has been estimated to range from 8 to 20 cm2, which approximates the size of the palm of an adult hand.17 Bacteremia and endotoxemia can be induced by dental procedures, as well as by usual daily activities (such as chewing), leading to an elevated inflammatory state and stimulating increases in the levels of serum inflammatory markers.1,12,18 Thus, locally produced proinflammatory mediators, such as interleukin-1 (IL-1), IL-6, tumour necrosis factor alpha (TNF-α) and prostaglandin E2 (PGE2), move into the systemic circulation and may subsequently exert effects on distant organ systems, as would be the case with other chronic infections or inflammatory processes and resulting in an acute-phase response. Elevated levels of these serum markers and mediators of inflammation have been observed in individuals with periodontitis.10 Moreover, patients with periodontitis, particularly those with gram-negative organisms such as Porphyromonas gingivalis, Tannerella forsythia and Prevotella intermedia, have significantly higher levels of C-reactive protein (CRP) and fibrinogen than those without periodontitis.19 Periodontal treatment not only reduces clinically evident inflammation, but also has been associated with decreases in IL-6, TNF-α and CRP, indicating that periodontal diseases have systemic effects extending beyond the local periodontal environment.20
Chronic inflammation through the action of inflammatory mediators is mainly associated with the development of insulin resistance, which is influenced by genetically modified environmental factors, including decreased physical activity, poor nutrition, obesity and infection.7,21 In the obesity-related model of the development of insulin resistance, activated adipocytes release abnormal levels of bioactive molecules, such as lipids, fatty acids, monocyte chemoattractant protein-1 and various inflammatory mediators (e.g., CRP, plasminogen activator inhibitor-1, TNF-α and IL-6). The release of these cytokines and other mediators results in the local recruitment of monocytes within the adipose tissues. With differentiation of the monocytes into macrophages comes an increase in the release of inflammatory factors and chemokines locally within the adipose tissue but also systemically, such that the inflammatory response is propagated to various tissues, especially to insulin-sensitive organs such as the liver and skeletal muscle, thus contributing to overall insulin resistance.22 One of the earliest studies to link the release of inflammatory substances from adipose tissues to insulin resistance in type 2 diabetes showed that TNF-α mRNA and protein were induced locally within adipose tissue and systemically in the plasma (see details in Figs. 1 and 3). When the expression of TNF-α was inhibited in a rodent model (fa/fa) by use of a recombinant TNF-α receptor–immunoglobulin G chimeric protein, insulin sensitivity improved, which suggested that this cytokine has a direct role in the development of insulin resistance.23 Thus, a mechanism was proposed that links the expression of TNF-α and other inflammatory mediators to the development of insulin resistance in obesity and type 2 diabetes.22 In this model, receptor ligands, such as inflammatory cytokines, bacterial lipopolysaccharides, lipids, free fatty acids, other microbial products and advanced glycation end products, activate the intracellular pathways, such as the I-kappa-B (IκB), I-kappa-B kinase-β (IKKβ), nuclear factor-kappa B (NF-κβ) and the protein c-Jun N-terminal kinase (JNK) axes. JNK has been shown to promote insulin resistance through the phosphorylation of serine residues in the insulin receptor substrate-1. Insulin receptor signalling, which normally occurs through a tyrosine kinase cascade, is inhibited by counter-regulatory phosphorylation of serine and threonine. Unlike JNK, IKKβ causes insulin resistance through transcriptional activation of NF-κB. This protein transcription factor is known to initiate the transcription of a variety of genes for compounds involved in insulin resistance, such as the genes for cytokines (TNF-α, IL-1, IL-6 and IL-8), growth factors, adhesion molecules and acute phase proteins. Activation of IKKβ leads to the phosphorylation of IκB, a cytosolic inhibitor of NF-κB. Phosphorylation targets IκB for ubiquitination and proteasomal degradation, freeing NF-κB to translocate to the nucleus where it regulates the transcription of target genes promoting insulin resistance. Other cellular stressors may activate these pathways, such as protein kinase C activators and oxidants. Once activated in the tissues, especially in the adipose tissue and associated immune cells, these processes may become self-perpetuating through a positive feedback loop created by the proinflammatory cytokines. 22
Given these mechanisms promoting insulin resistance, it seems that in individuals with type 2 diabetes and periodontitis, an elevated chronic systemic inflammatory state induced by periodontal disease may contribute to insulin resistance through a “feed-forward” mechanism, worsening glycemic control1,12 (Fig. 3). This might explain why periodontitis increases the risk of poor glycemic control among patients with type 2 diabetes.5 Periodontitis may also contribute to the elevation of serum inflammation mediators through enhanced in vitro production of TNF-α, IL-1β and PGE2 by monocytes, as has been shown in patients with both diabetes and periodontitis. This may indicate an innate hyperresponsiveness of these monocytes to periodontal bacterial challenge.24,25 Periodontitis may also play a role through the translocation of gram-negative species and their products from the periodontal biofilm into the circulation18,25 and through direct cytokinemia from the gingival crevicular fluid (i.e., translocation of cytokines from the periodontal space into the circulation).25 With regard to the last of these mechanisms, poorer glycemic control was associated with increased levels of cytokines, especially IL-1β, in the gingival crevicular fluid.26 In individuals with type 2 diabetes and periodontitis, serum levels of TNF-α were significantly correlated with the severity of periodontal destruction, plasma endotoxin and IL-1β levels in the gingival crevicular fluid, but not with body mass index (BMI), serum glucose or hemoglobin A1c (HbA1c) levels. Furthermore, there was a dose–response relationship between the severity of periodontitis and serum TNF-α levels, which suggested that periodontal disease may play a major role in elevating levels of this cytokine, which is closely linked to insulin resistance.25 An examination of NHANES III data from participants without diabetes revealed a positive association between BMI and clinical attachment loss. Moreover, those in the highest quartile of body mass (BMI ≥ 30.8 kg/m2) had significantly higher serum levels of TNF-α and soluble TNF-α receptors than those in the lowest quartile of body mass (BMI < 24.6 kg/m2). These data suggest that obesity is associated with both systemic inflammation and periodontal disease and that insulin resistance may mediate this relationship.27
Impact of Periodontal Treatment on Systemic Inflammatory State and Glycemic Control
Periodontal treatment that reduces periodontal inflammation may help to restore insulin sensitivity, thereby improving glycemic control.1,12 Intervention studies showing a decrease in the level of systemic inflammatory markers and improved glycemic control following periodontal therapy would support such a hypothesis. Studies of patients with both diabetes and periodontitis have shown that nonsurgical periodontal therapy with adjunctive local delivery of minocycline reduced circulating levels of TNF-α.28,29 In one of those studies, the reduction in serum levels of TNF-α was accompanied by, and strongly correlated with, a significant decrease in mean HbA1c values (from 8% to 7.1%).28 Conversely, a pilot study showed that serum levels of TNF-α were not significantly affected 4 weeks after mechanical periodontal therapy.30 In the same study, systemic levels of mediators such as CRP and soluble E-selectin were significantly reduced following nonsurgical periodontal debridement.30
Outcomes of a meta-analysis of 10 intervention trials involving 456 patients with diabetes (type 1 or type 2) showed that following mechanical periodontal debridement, HbA1c levels decreased by an average of 0.38% over all studies, by 0.66% among patients with type 2 diabetes and by 0.71% among cases in which antibiotics were administered. However, none of these changes were statistically significant.31 A recent single-blind, randomized controlled trial confirmed the results of the meta-analysis, showing that periodontal therapy combined with diabetes medication had no statistically significant effect on levels of HbA1c relative to no treatment.32 Other studies have shown significant improvements in glycemic control with periodontal therapy.33,34 These conflicting data are difficult to interpret because of the wide range of medical treatment regimens used in study populations, inadequate sample sizes, combined enrolment of patients with type 1 and type 2 diabetes, confounding by smoking and BMI, and study design (e.g., studies examining only short-term outcomes or pilot studies). Although the 0.7% improvement in HbA1c levels attributed to mechanical periodontal debridement and antibiotic therapy reported in the meta-analysis was not statistically significant, its clinical significance should not be minimized, given that the less potent class of oral glucose-lowering agents, the α-glucosidase inhibitors, reduces HbA1c level by 0.5% to 1%.35 Other classes of oral agents, such as insulin secretagogues, biguanides and thiazolidinediones, as well as nutritional therapy and physical activity, improve glycemic control to a similar degree, with 1% to 2% reduction in HbA1c.35 Therefore, since periodontal treatment appears to have the same power to lower HbA1c as other glucose-lowering therapies, it may represent an alternative or adjunctive therapy for improving insulin sensitivity and glycemic control in patients with both type 2 diabetes and periodontitis.
Final Considerations
As this literature review has indicated, the cytokine-induced inflammatory state in periodontitis can contribute to the overall low-grade inflammation that occurs in diabetes. This low-grade inflammation is characterized by chronic activation of the patient’s innate immunity and, consequently, may aggravate insulin resistance and adversely affect glycemic control. Current evidence is conflicting, but does support, to some extent, the hypothesis that periodontal treatment may restore insulin sensitivity and improve glycemic control by reducing periodontal inflammation and serum levels of cytokines and inflammatory markers. Further research is required to clarify this aspect of how periodontal diseases influence diabetes. As scientific knowledge in this area accumulates, the clinician must determine its relevance to patient care. Nonetheless, information about host responses and modulation factors in diabetes, periodontitis and diabetes-associated periodontitis may be used for therapeutic purposes. As our understanding of these diseases deepens, the focus is shifting from diagnosis and treatment to prevention and health promotion. Many cases of diabetes may remain undiagnosed, and opportunistic screening for diabetes in the dental office, based on self-reported data and clinical periodontal parameters, might be effective in identifying some of these cases. Active and supportive therapy to improve insulin sensitivity and glycemic control, such as preventing the recurrence of periodontal disease and tooth mortality in patients with diabetes, should be considered important components of treatment. As evidence of the close link between inflammatory periodontal diseases and diabetes continues to accumulate, physicians and oral health professionals should interact to a greater extent, to improve general health care and glycemic control and to prevent complications among patients with diabetes.
THE AUTHORS
|
Dr. Tunes is an assistant professor in the division of periodontics, School of Dentistry of Bahiana Medical School and Public Health (EBMSP), Salvador, Brazil. |
|
|
Dr. Foss-Freitas is an associate professor in the division of endocrinology and metabolism, department of medical clinics, Medical School of Ribeirão Preto, University of São Paulo, Ribeirão Preto, Brazil. |
|
|
Dr. Nogueira-Filho is an associate professor in the department of dental diagnostics and surgical sciences and director of undergraduate periodontics, faculty of dentistry, University of Manitoba, Winnipeg, Manitoba. |
|
Correspondence to: Dr. Getúlio Nogueira, D344-790 Bannatyne Ave, Winnipeg, MB R3E 0W2. Email: nogueira@cc.umanitoba.ca
The authors have no declared financial interests.
This article has been peer reviewed.
References
- Mealey BL, Oates TW. Diabetes mellitus and periodontal diseases. J Periodontol. 2006;77:1289-303.
- Lipscombe LL, Hux JE. Trends in diabetes prevalence, incidence, and mortality in Ontario, Canada 1995-2005: a population-based study. Lancet. 2007;369(9563):750-6.
- Horn OK, Jacobs-Whyte H, Ing A, Bruegl A, Paradis G, Macaulay AC. Incidence and prevalence of type 2 diabetes in the First Nation community of Kahnawá:ke, Quebec, Canada, 1986-2003. Can J Public Health. 2007;98(6):438-43.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet. 1998;352:837-53.
- Taylor GW. Bidirectional interrelationships between diabetes and periodontal diseases: an epidemiologic perspective. Ann Periodontol. 2001;6:99-112.
- Löe H. Periodontal disease. The sixth complication of diabetes mellitus. Diabetes Care. 1993;16(Suppl.1):329-34.
- Pickup JC, Hil DP, Path FRC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 2004;27:813-23.
- Spranger J, Kroke A, Möhlig M, Hoffmann K, Bergmann MM, Ristow M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes. Results of the prospective population-based European prospective investigation into cancer and nutrition (EPIC)-Potsdam Study. Diabetes. 2003;52:812-7.
- Nassar H, Kantarci A, Van Dyke TE. Diabetes periodontitis: a model for activated innate immunity and impaired resolution of inflammation. Periodontol 2000. 2007;43:233-44.
- Loos BG. Systemic markers of inflammation in periodontitis. J Periodontol. 2005;76:2106-15.
- Salvi GE, Carollo-Bittel B, Lang NP. Effects of diabetes mellitus on periodontal and peri-implant conditions. Update on association and risks. J Clin Periodontol. 2008;35(Suppl.8):398-409.
- Mealey BL, Rose LF. Diabetes mellitus and inflammatory periodontal diseases. Curr Opin Endocrinol Diabetes Obes. 2008;15:135-41.
- Solskolne WA, Kingler A. The relationship between periodontal diseases and diabetes: an overview. Ann of Periodontol. 2001;6:91-8.
- Saito T, Shimazaki Y, Kiyohara Y, Kato I, Kubo M, Iida M, et al. The severity of periodontal disease is associated with the development of glucose intolerance in non-diabetics: the Hisayama study. J Dent Res. 2004;83:485-90.
- Shimazaki Y, Saito T, Yonemoto K, Kiyohara Y, Iida M, Yamashita, Y. Relationship of metabolic syndrome to periodontal disease in Japanese women: the Hisayama study. J Dent Res. 2007;86:271-5.
- Jansson H, Lindholm E, Lindh C, Groop L, Bratthall G. Type 2 diabetes and risk for periodontal disease: a role for dental health awareness. J Clin Periodontol. 2006;33:408-14.
- Hujoel PP, White BA, Garcia RI, Listgarten MA. The dentogingival epithelial surface area revisited. J Periodontal Res. 2001;36:48-55.
- Geerts SO, Nys M, De Mol P, Charpentier J, Albert A, Legrand V, et al. Systemic release of endotoxins induced by gentle mastication: association with periodontitis severity. J Periodontol. 2002;73:73-8.
- Noack B, Genco RJ, Trevisan M, Grossi S, Zambon JJ, De Nardin E. Periodontal infections contribute to elevated systemic C-reactive protein level. J Periodontol. 2001;72:1221-7.
- D’Aiuto F, Parkar M, Andreou G, Suvan J, Brett PM, Ready D, et al. Periodontitis and systemic inflammation: control of the local infection is associated with a reduction in serum inflammatory markers. J Dent Res. 2004;83:156-60.
- King GL. The role of inflammatory cytokines in diabetes and its complications. J Periodontol. 2008;79:1527-34.
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793-801.
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87-91.
- Salvi GE, Beck JD, Offenbacher S. PGE2, IL-1β, and TNF-α responses in diabetics as modifiers of periodontal disease expression. Ann Periodontol. 1998;3:40-50.
- Engebretson S, Chertog R, Nichols A, Hey-Hadavi J, Celenti R, Grbic J. Plasma levels of tumour necrosis factor-α in patients with chronic periodontitis and type 2 diabetes. J Clin Periodontol. 2007;34:18-24.
- Engebretson SP, Hey-Hadavi J, Ehrhardt FJ, Hsu D, Celenti RS, Grbic JT, et al. Gingival crevicular fluid levels of interlukin-1beta and glycemic control in patients with chronic periodontitis and type 2 diabetes. J Periodontol. 2004;75:1203-8.
- Genco RJ, Grossi SG, Ho A, Nishimura F, Murayama Y. A proposed model linking inflammation to obesity, diabetes, and periodontal infections. J Periodontol. 2005;76:2075-84.
- Iwamoto Y, Nishimura F, Nakagawa M, Sugimoto H, Shikata K, Makino H, et al. The effect of antimicrobial periodontal treatment on circulating tumor necrosis factor-alpha and glycated hemoglobin level in patients with type 2 diabetes. J Periodontol. 2001;72:774-8.
- Nishimura F, Iwamoto Y, Mineshiba J, Shimizu A, Soga Y, Murayama Y. Periodontal disease and diabetes mellitus: the role of tumor necrosis factor–alpha in a 2-way relationship. J Periodontol. 2003;74:97-102.
- Lalla E, Kaplan S, Yang J, Roth GA, Papapanou PN, Greenberg S. Effects of periodontal therapy on serum C-reactive protein, sE-selectin and tumor necrosis factor-alpha secretion by peripheral blood-derived macrophages in diabetes. A pilot study. J Periodontal Res. 2007;42:274-82.
- Janket SJ, Wightman A, Baird AE, Van Dyke TE, Jones JA. Does periodontal treatment improve glycemic control in diabetes patients? A meta-analysis of intervention studies. J Dent Res. 2005;84:1154-9.
- Jones JA, Miller DR, Wehler CJ, Rich SE, Krall-Kaye EA, McCoy LC, et al. Does periodontal care improve glycemic control? The department of veterans’ affairs dental diabetes study. J Clin Periodontol. 2007;34:46-52.
- Kiran M, Arpak N, Ünsal E, Erdogan MF. The effect of improved periodontal health on glycemic control in type 2 diabetes mellitus. J Clin Periodontol. 2005;32:266-72.
- Faria-Almeida R, Navarro A, Bascones A. Clinical and metabolic changes after conventional treatment of type 2 diabetes patients with chronic periodontitis. J Periodontol. 2006;77:591-8.
- Powers AC. Diabetes mellitus. In: Jameson JL, Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, editors. Harrison’s endocrinology & metabolism, 16th ed. Philadelphia [Pennsylvania]: The McGraw-Hill Companies, Inc.; 2006. p. 283-331.