Un garçon de 9 ans a été dirigé par son dentiste vers le département de médecine dentaire de l'hôpital Riuniti di Bergamo pour une évaluation et un éventuel traitement orthodontique. Son syndrome avait été diagnostiqué durant un examen ophtalmologique réalisé lorsque l'enfant avait 3 ans. Sur le plan clinique, le patient présentait une exophtalmie (ill. 1) et un léger hypertélorisme.
L'examen intrabuccal a révélé une grave hypodontie et des dents microdontiques – deux manifestations typiques de ce syndrome. La dentition était mixte (ill. 2 et 3, tableau 1).
L'examen radiographique a montré que 10 dents permanentes étaient absentes, soit 6 au maxillaire supérieur et 4 au maxillaire inférieur (ill. 4).
Le tracé et l'analyse céphalométriques indiquaient un surplomb vertical squelettique trop grand et un schéma facial normodivergent avec birétrusion. Le patient présentait également un diastème central et une asymétrie dentaire.
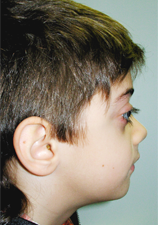 Ill. 1 : Vue de profil du patient présentant une exophtalmie.
Ill. 1 : Vue de profil du patient présentant une exophtalmie.
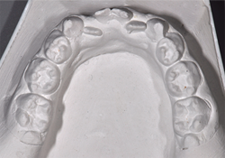 Ill. 2 : Modèle en plâtre de la dentition inférieure.
Ill. 2 : Modèle en plâtre de la dentition inférieure.
 Ill. 3 : Modèle en plâtre de la dentition supérieure.
Ill. 3 : Modèle en plâtre de la dentition supérieure.
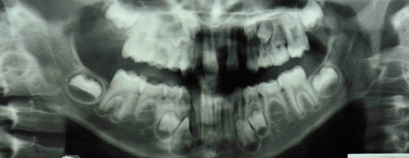 Ill. 4 : Radiographie panoramique montrant les dents n'ayant pas fait éruption et les dents permanentes absentes.
Ill. 4 : Radiographie panoramique montrant les dents n'ayant pas fait éruption et les dents permanentes absentes.
Tableau 1 Tableau illustrant l'état des dents du patient
| Dents permanentes absentes |
18
48 |
15
45 |
12 | 22 |
25
35 |
28
38 |
||||||||||
| Dents n'ayant pas fait éruption |
17
47 |
14
44 |
13
43 |
23
33 |
24
34 |
27
37 |
||||||||||
| Dents présentes |
16
46 |
55
85 |
54
84 |
53
83 |
52
42 |
11
41 |
21
31 |
32 |
63
73 |
64
74 |
65
75 |
26
36 |
De quelle affection s'agit-il?
Discussion
Le syndrome de Rieger est une affection autosomique dominante rare qui englobe un éventail d'anomalies du développement de la chambre antérieure de l'œil. Ces anomalies s'accompagnent d'un risque de 50 % de glaucome – un état habituellement réfractaire au traitement médical qui peut mener à la cécité. Lorsque d'autres anomalies accompagnent l'anomalie oculaire, notamment des anomalies somatiques craniofaciales, dentaires et du développement, on parle du syndrome d'Axenfeld-Rieger. Il a été démontré que deux gènes, tous deux des facteurs de transcription (Pitx2 à 4q25 et Foxc1 à 6p25), sont responsables du phénotype de ce syndrome1,2 dont la prévalence a été estimée à 1 pour 200 000 habitants.
Les manifestations craniofaciales incluent une circonférence occipito-frontale normale, un large front, des anomalies de la selle turcique3, l'hypertélorisme, des fentes palpébrales obliques vers le bas, une voûte nasale plate, un nez court et relevé, un sillon sous-nasal mal défini, un sous-développement des maxillaires, une mince lèvre supérieure, une position bouche ouverte, des oreilles basses, une hernie ombilicale ou une peau péri-ombilicale redondante et, dans de rares cas, de multiples anomalies congénitales.
Les anomalies dentaires sont considérées comme une manifestation essentielle du syndrome d'Axenfeld-Rieger. Les anomalies buccales incluent un frein labial supérieur hyperplasique, des dents antérieures en forme de cône, une microdontie et une hypodontie (en particulier des dents antérieures supérieures, primaires et permanentes). Les incisives et les canines supérieures sont les dents les plus souvent absentes, alors que les deuxièmes prémolaires le sont parfois. Parmi les autres anomalies dentaires possibles, mentionnons une hypoplasie de l'émail, des dents de forme conique, un raccourcissement des racines, le taurodontisme et le retard d'éruption4.
Il est important d'assurer un diagnostic précoce de ce syndrome sur la base des manifestations dentofaciales et systémiques, car ceci peut permettre de prévenir les complications oculaires subséquentes.
Il s'agit d'un trouble du développement héréditaire à expression variable et à pénétrance complète. Dans 30 % des cas, cet état est le résultat de mutations de novo et, dans 70 % des cas, l'hérédité est autosomique dominante5.
Les patients atteints du syndrome d'Axenfeld-Rieger ont souvent des anomalies craniofaciales avec involution cutanée péri-ombilicale. Ces signes peuvent être associés à une grande variété d'autres manifestations, notamment une insuffisance staturale, des anomalies des membres, une selle turcique vide, des anomalies apophysaires et une variété de troubles neurologiques et dermatologiques. Le diagnostic différentiel devrait inclure le nanisme, l'hypopituitarisme et un déficit en hormone de croissance.
Les manifestations oculaires du syndrome d'Axenfeld-Rieger incluent l'hypoplasie du stroma irien, l'ectropion de l'uvée, la corectopie, des défauts pleine épaisseur de l'iris, une grave atrophie de l'iris et d'importantes synéchies antérieures périphériques. Le glaucome se manifeste dans 50 % des cas, habituellement tôt durant l'enfance ou l'âge adulte, à la suite d'un angle d'anomalie connexe ou de la fermeture secondaire de l'angle par des synéchies6.
Selon Childers et Wright7, le syndrome d'Axenfeld-Rieger est attribuable à des lésions ou à la migration anormale des cellules de la crête neurale, le mésostroma ectodermique, tard durant la gestation. Ces cellules donnent normalement naissance à du tissu conjonctif, de l'os et du cartilage et sont nécessaires au développement des structures craniofaciales, oculaires et dentaires. Des aberrations dans le développement de la crête neurale pourraient expliquer les anomalies morphologiques de l'hypoplasie des maxillaires supérieur et inférieur, l'hypertélorisme, les anomalies dentaires et le syndrome de la selle turcique vide.
Ozeki et ses collègues8 ont mené une étude qui a permis d'élucider le lien entre les troubles oculaires et d'autres manifestations cliniques. Ils ont évalué 21 patients âgés de 1 mois à 41 ans atteints du syndrome d'Axenfeld-Rieger. Tous les patients présentaient un glaucome, bilatéral dans 17 cas et unilatéral dans 4 cas. Le glaucome était associé à des anomalies dentaires dans 9 cas, à des anomalies faciales dans 5 cas et au syndrome d'Alagille dans 3 cas.
Les patients atteints du syndrome d'Axenfeld-Rieger doivent subir des examens ophtalmologiques régulièrement durant toute leur vie pour surveiller les changements dans le disque du nerf optique et la pression intra-oculaire. Comme ce syndrome est souvent familial, les personnes consanguines devraient aussi être suivies. Plus de 50 % des patients atteints de ce syndrome manifestent un glaucome secondaire, ce qui augmente le risque de perte de vision et de cécité. Le traitement initial du glaucome est médical et consiste en l'application topique de bêtabloquants, d'inhibiteurs de l'anhydrase carbonique ou d'alpha-agonistes pour réduire la pression intra-oculaire. Une intervention chirurgicale (p. ex., goniotomie, trabéculectomie et trabéculotomie) est nécessaire chez la plupart des patients. La trabéculectomie avec antimétabolite est le traitement de choix pour les patients atteints du syndrome d'Axenfeld-Rieger9,10.
Traitement
Il est important que les dentistes connaissent le syndrome d'Axenfeld-Rieger, car les manifestations dentaires et faciales peuvent en être les premiers symptômes perceptibles. Un diagnostic et des interventions précoces sont nécessaires pour prévenir les complications oculaires.
Notre patient est toujours suivi pour un traitement orthodontique. Le but de ce traitement est de préserver les dents primaires et de résoudre les problèmes de chevauchement afin de maintenir la fonction et l'esthétique. À long terme, les implants dentaires constituent le traitement le plus probable pour ces patients.
Les patients atteints du syndrome d'Axenfeld-Rieger ont de multiples problèmes et requièrent une démarche interdisciplinaire, laquelle est jugée essentielle pour assurer le meilleur traitement et suivi.
LES AUTEURS
|
|
Le Dr Villa est fellow à l'École de médecine dentaire de Harvard et résident dans la Division de la médecine buccale et dentaire à l'Hôpital Brigham and Women's de Boston au Massachusetts. Il était fellow au Département de médecine dentaire à l'Hôpital Riuniti di Bergamo, Bergamo, Italie, au moment de la rédaction de cet article. |
|
|
La Dre Albonico est dentiste traitant au Département de médecine dentaire, l'Hôpital Riuniti di Bergamo, Bergamo, Italie. |
|
|
Le Dr Villa exerce dans un cabinet privé à Bergamo (Italie). Il était chef du Département de médecine dentaire à l'Hôpital Riuniti di Bergamo, Bergamo, Italie, au moment de la rédaction de cet article. |
Écrire au : Dr Alessandro Villa, Division de la médecine buccale et dentaire, Hôpital Brigham and Women's, 1620, rue Tremont, Pièce BC-3-028 Boston, MA 02120. Courriel : avilla@partners.org
Les auteurs n'ont aucun intérêt financier déclaré.
Cet article a fait l'objet d'une révision par des pairs.
Références
- Lines MA, Kozlowski K, Walter MA. Molecular genetics of Axenfeld-Rieger malformations. Hum Mol Genet. 2002;11(10):1177-84.
- Weisschuh N, Wolf C, Wissinger C, Gramer E. A novel mutation in the FOXC1 gene in a family with Axenfeld-Rieger syndrome and Peters' anomaly. Clin Genet. 2008;74(5):476-80. Epub 2008 May 21.
- Meyer-Marcotty P, Weisschuh N, Dressler P, Hartmann J, Stellzig-Eisenhauer A. Morphology of the sella turcica in Axenfeld-Rieger syndrome with PITX2 mutation. J Oral Pathol Med. 2008;37(8):504-10. Epub 2008 Mar 10.
- Brooks JK, Coccaro PJ, Zarbin MA. The Rieger anomaly concomitant with multiple dental, craniofacial, and somatic midline anomalies and short stature. Oral Surg Oral Med Oral Pathol. 1989;68(6):717-24.
- Grosso S, Farnetani MA, Berardi R, Vivarelli R, Vanni M, Morgese G, et al. Familial Axenfeld-Rieger anomaly, cardiac malformations, and sensorineural hearing loss: a provisionally unique genetic syndrome? Am J Med Genet. 2002;111(2):182-6.
- Gould DB, John SW. Anterior segment dysgenesis and the developmental glaucomas are complex traits. Hum Mol Genet. 2002; 11(10):1185-93.
- Childers NK, Wright JT. Dental and craniofacial anomalies of Axenfeld-Rieger syndrome. J Oral Pathol. 1986;15(10):534-9.
- Ozeki H, Shirai S, Ikeda K, Ogura Y. Anomalies associated with Axenfeld-Rieger syndrome. Graefes Arch Clin Exp Ophthalmol 1999;237(9):730-4.
- O'Dwyer EM, Jones DC. Dental anomalies in Axenfeld-Rieger syndrome. Int J Paediatr Dent. 2005;15(6):459-63.
- Shields MB. Axenfeld-Rieger and iridocorneal endothelial syndromes: two spectra of disease with striking similarities and differences. J Glaucoma. 2001;10(5 Suppl 1):S36-8.